Variant sorting screen tutorial¶
GWAS variant screen with per-variant gRNA tiling design, selected based on FACS signal quantiles.
| Library design | Variant (gRNAs tile each target variant) 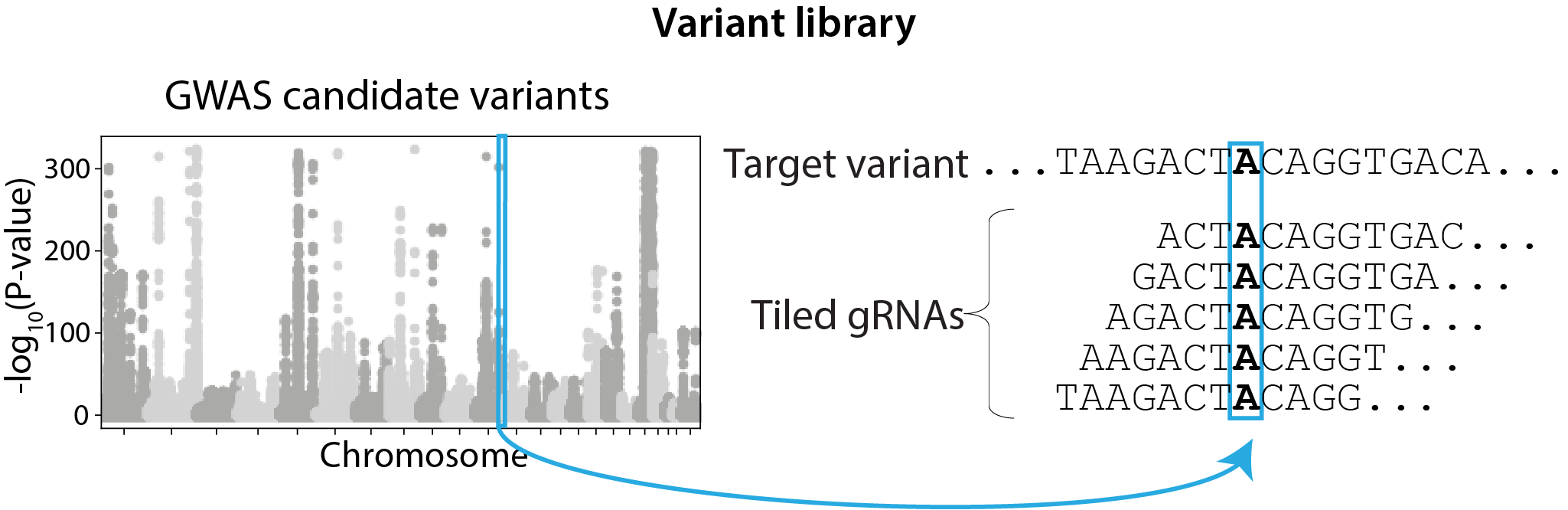 |
|---|---|
| Selection | Cells are sorted based on FACS signal quantiles 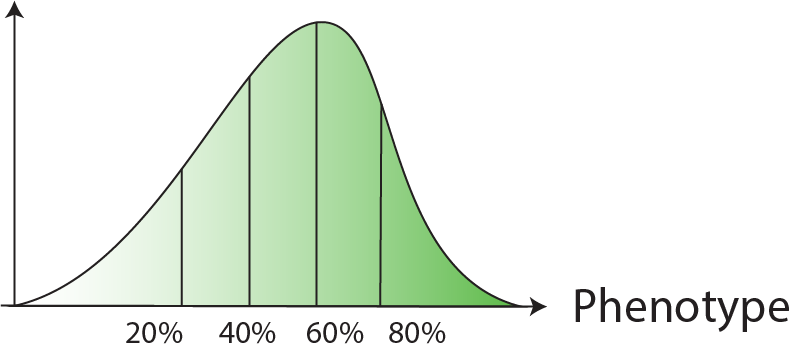 |
Example workflow¶
screen_id=var_mini_screen
working_dir=tests/data/
# 1. Count gRNA & reporter
bean count-samples \
--input ${working_dir}/sample_list.csv `# Contains fastq file path; see test file for example.`\
-b A `# Base A is edited (into G)` \
-f ${working_dir}/test_guide_info.csv `# Contains gRNA metadata; see test file for example.`\
-o ./ `# Output directory` \
-r `# Quantify reporter edits` \
-n ${screen_id} `# ID of the screen to be counted`
# count-samples output from above test run is too low in read depth. Downstream processes can be run with test file included in the Github repo.
# (Optional) Profile editing patterns
bean profile tests/data/${screen_id}.h5ad --pam-col '5-nt PAM'
# 2. QC samples & guides
bean qc \
${working_dir}/bean_count_${screen_id}.h5ad `# Input ReporterScreen .h5ad file path` \
-o ${working_dir}/bean_count_${screen_id}_masked.h5ad `# Output ReporterScreen .h5ad file path` \
-r ${working_dir}/qc_report_${screen_id} `# Prefix for QC report` \
-b ` # Remove replicates with no good samples.
# 3. Quantify variant effect
bean run sorting variant \
${working_dir}/${screen_id}_masked.h5ad \
-o ${working_dir}/ \
--fit-negctrl \
--scale-by-acc \
--acc-bw-path tests/data/accessibility_signal_chr6.bw
See more details below.
1. Count gRNA & reporter (bean count-samples)¶
screen_id=my_sorting_tiling_screen
# 1. Count gRNA & reporter
bean count-samples \
--input ${working_dir}/sample_list.csv `# Contains fastq file path; see test file for example.`\
-b A `# Base A is edited (into G)` \
-f ${working_dir}/test_guide_info.csv `# Contains gRNA metadata; see test file for example.`\
-o ./ `# Output directory` \
-r `# Quantify reporter edits` \
-n ${screen_id} `# ID of the screen to be counted`
Make sure you follow the input file format for seamless downstream steps. This will produce ./bean_count_${screen_id}.h5ad.
(Optional) Profile editing pattern (bean profile)¶
You can profile the pattern of base editing based on the allele counts.
bean profile tests/data/${screen_id}.h5ad --pam-col '5-nt PAM'
Output¶
Output will be written under ${working_dir}/bean_profile.${screen_id}/. See example output here.
2. QC samples & guides (bean qc)¶
Base editing data will include QC about editing efficiency. As QC uses predefined column names and values, beware to follow the input file guideline, but you can change the parameters with the full argument list of bean qc. (Common factors you may want to tweak is --ctrl-cond=bulk and --lfc-conds=top,bot if you have different sample condition labels.)
bean qc \
${screen_id}.h5ad `# Input ReporterScreen .h5ad file path` \
-o ${screen_id}_masked.h5ad `# Output ReporterScreen .h5ad file path` \
-r qc_report_${screen_id} `# Prefix for QC report`
If the data does not include reporter editing data, you can provide --no-editing flag to omit the editing rate QC.
Output¶
Output will be written under ${working_dir}/. See example output here.
3. Quantify variant effect (bean run)¶
bean run can take 3 run options to quantify editing rate:
From reporter + accessibility
If your gRNA metadata table (${working_dir}/test_guide_info.csvabove) included per-gRNA accessibility score,bean run sorting variant \ ${working_dir}/${screen_id}_masked.h5ad \ -o ${working_dir}/ \ --fit-negctrl \ --scale-by-acc \ --acc-col accessibility
If your gRNA metadata table (
${working_dir}/test_guide_info.csvabove) included per-gRNA chromosome & position and you have bigWig file with accessibility signal,bean run sorting variant \ ${working_dir}/${screen_id}_masked.h5ad \ -o ${working_dir}/ \ --fit-negctrl \ --scale-by-acc \ --acc-bw-path accessibility.bw
From reporter, without accessibility
This assumes the all target sites have the uniform chromatin accessibility.
bean run sorting variant \ ${working_dir}/${screen_id}_masked.h5ad \ -o ${working_dir}/ \ --fit-negctrl
No reporter information, assume the same editing efficiency of all gRNAs.
Use this option if your data don’t have editing outcome information.bean run sorting variant \ ${working_dir}/${screen_id}_masked.h5ad \ -o ${working_dir}/ \ --fit-negctrl \ --uniform-edit
Output¶
Output will be written under ${working_dir}/. See example output here.
See Subcommands for the full details.
