bean run¶
bean run: Quantify variant effects¶
BEAN uses Bayesian network to incorporate gRNA editing outcome to provide posterior estimate of variant phenotype. The Bayesian network reflects data generation process. Briefly,
Cellular phenotype (either for cells are sorted upon for sorting screen, or log(proliferation rate)) is modeled as the Gaussian mixture distribution of wild-type phenotype and variant phenotype.
The weight of the mixture components are inferred from the reporter editing outcome and the chromatin accessibility of the loci.
Cells with each gRNA, formulated as the mixture distribution, is sorted by the phenotypic quantile to produce the gRNA counts.
For the full detail on modeling, see the model description.
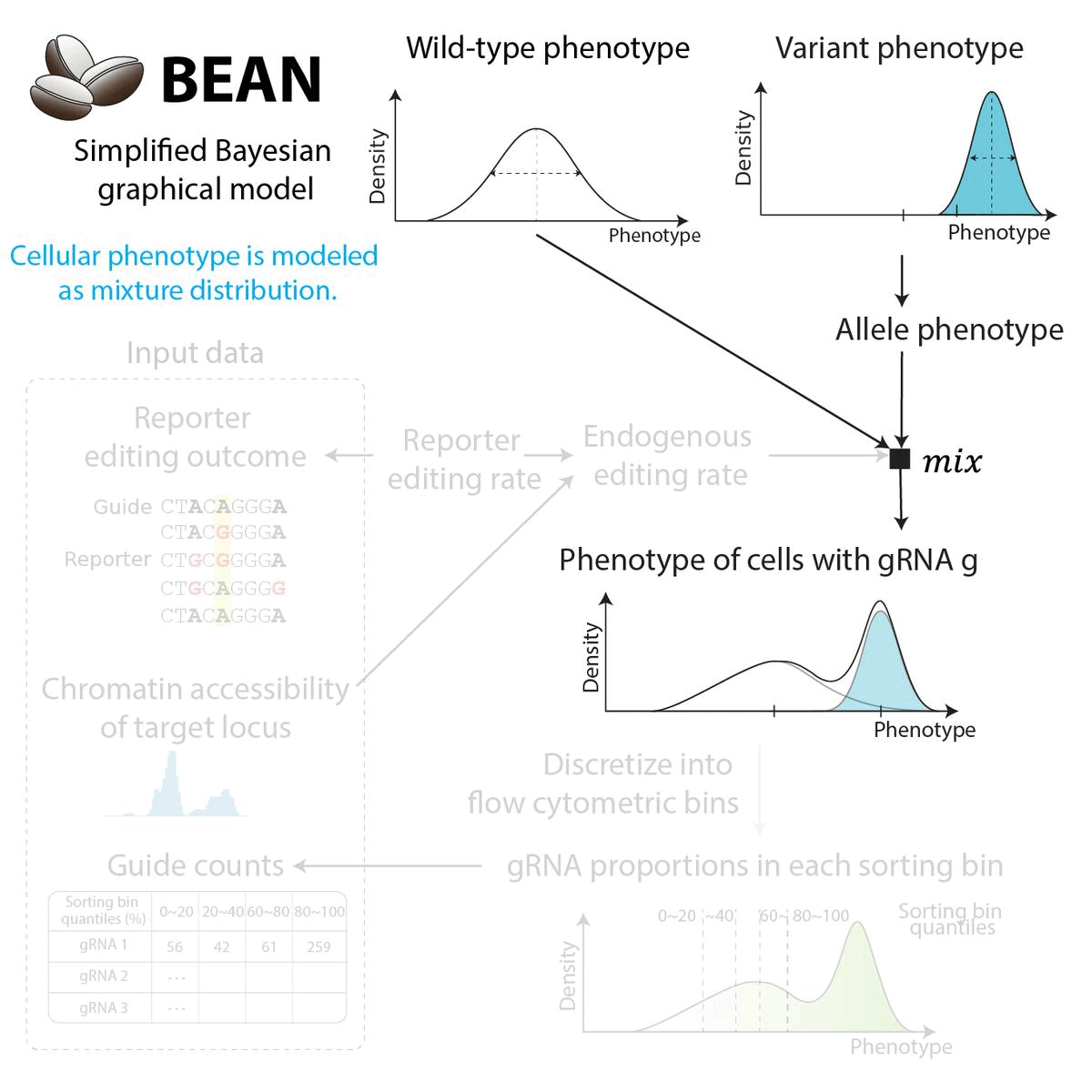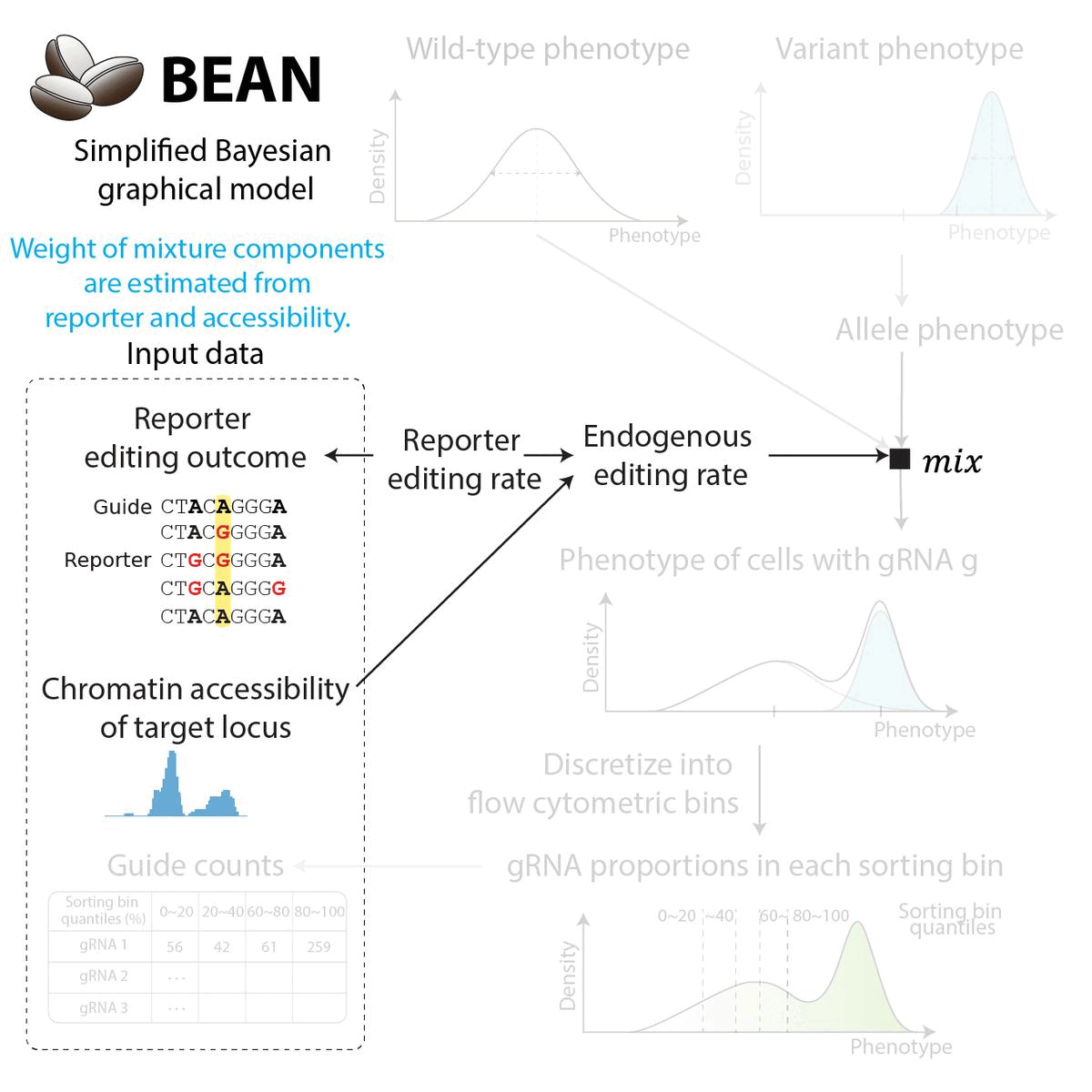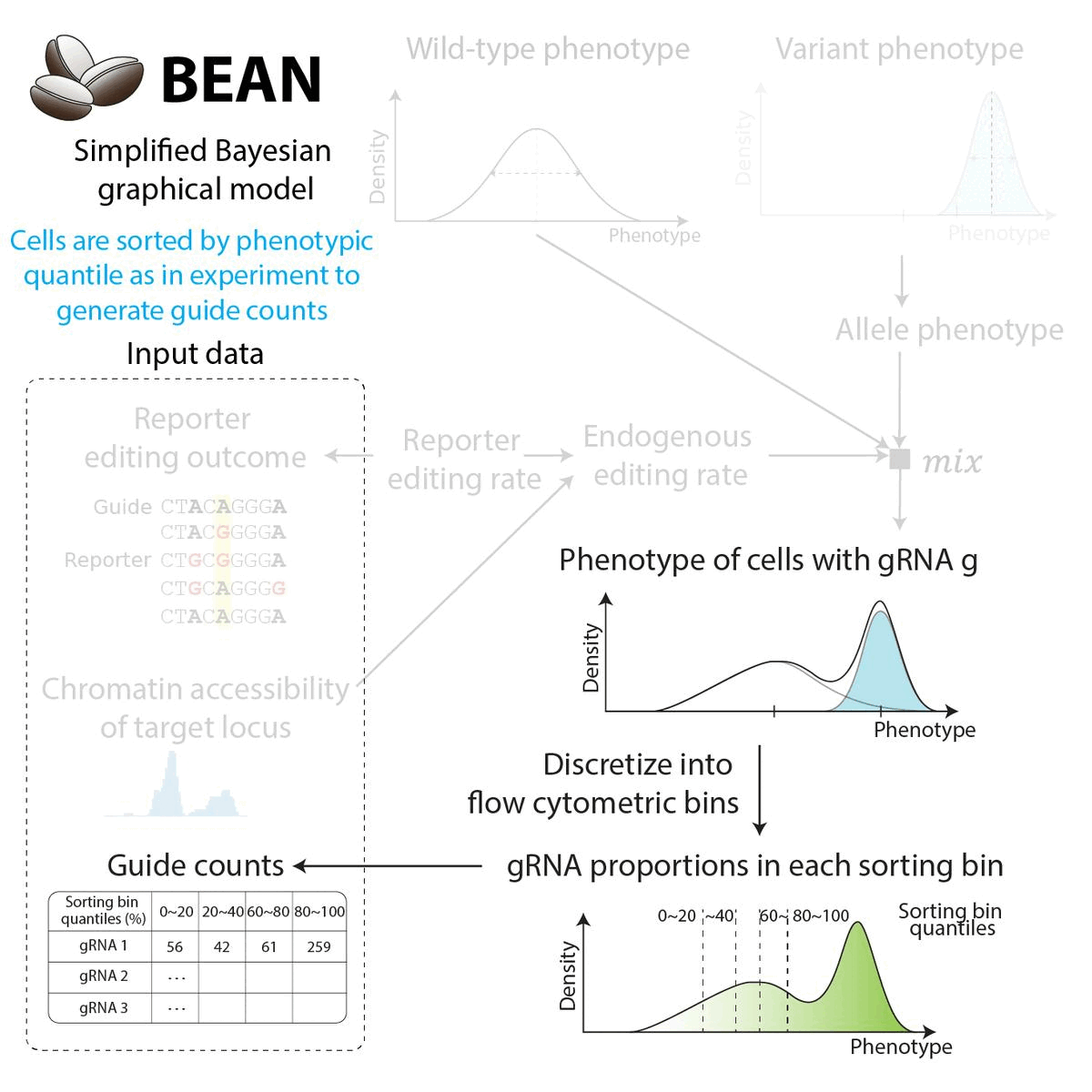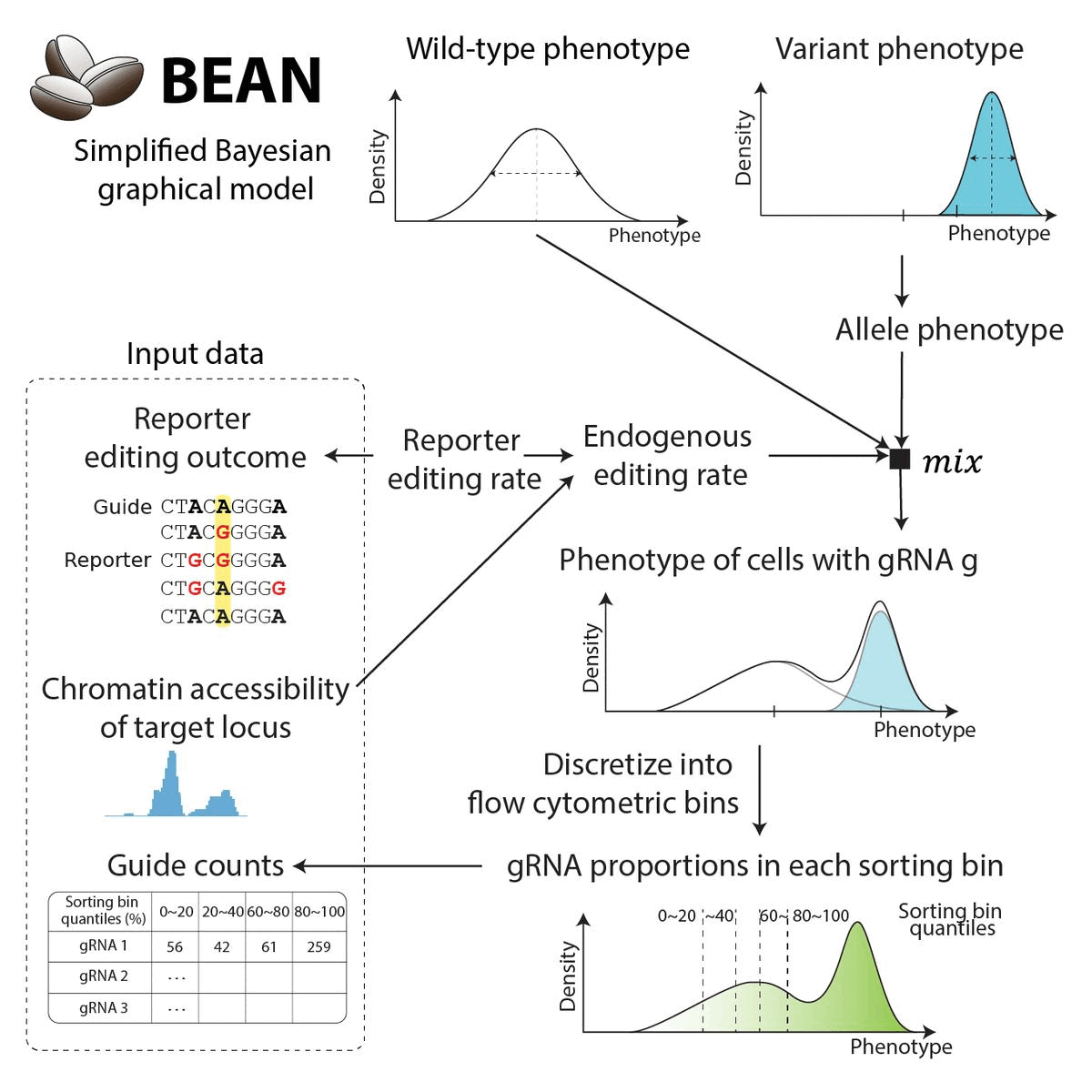
Usage example¶
bean run sorting[survival] variant[tiling] my_sorting_screen_filtered.h5ad \
[--uniform-edit, --scale-by-acc [--acc-bw-path accessibility_signal.bw, --acc-col accessibility]] \
-o output_prefix/ \
--fit-negctrl
See full list of parameters below.
Input¶
my_sorting_screen_filtered.h5ad can be produced by one of the following:
``bean count-samples` <(#bean-count-samples-count-reporter-screen-data>`_) when you have raw
.fastqfile(Limited to
bean run variantmode)bean create-screenwhen you have flat.csvtables of gRNA metadata table, sample metadata table, gRNA counts table (# guides x # samples), and optionally # edits table. .. code-block:: bashbean create-screen gRNA_info_table.csv sample_info_table.csv gRNA_counts_table.csv [–edits edit_counts_table.csv -o output.h5ad]
gRNA_info_table.csvshould have following columns.name: gRNA ID columntarget: This column denotes which target variant/element of each gRNA.target_group [Optional]: If negative control gRNA will be used, specify as “NegCtrl” in this column.
sample_info_table.csvshould have following columns.sample_id: ID of sequencing samplereplicate: Replicate # of this samplebin: Name of the sorting binupper_quantile: FACS sorting upper quantilelower_quantile: FACS sorting lower quantile
gRNA_counts_table.csvshould be formatted as follows.Columns include one of
sample_idcolumns insample_info_table.csvfile.1st row (row index) follows
name(gRNA ID) ingRNA_info_table.csvfile.
You can manually create the
AnnDataobject with more annotations including allele counts: see API tutorial for full detail.
Output¶
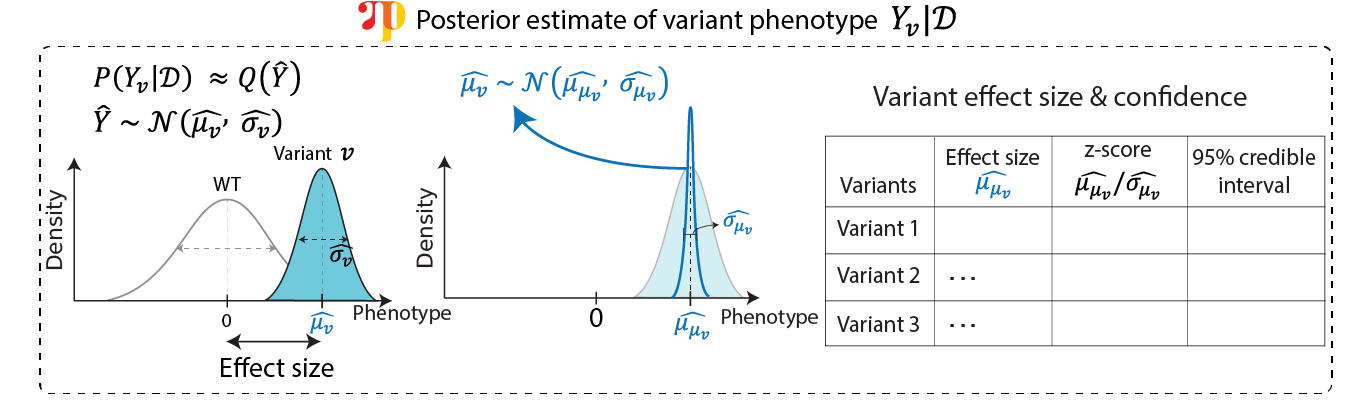
Above command produces
bean_element_result.[model_type].csv¶
Variant ID / grouping
edit: Variant ID.group: The grouping of the coding variants, assigned as one of nonsense/missense/synonymous.int_pos: The integer position of the noncoding variants.chrom: The chromosome of the variant.pos: The position of the variant. If coding variant, starts withAand the position specified 1-based amino acid position. If noncodig variant, numeric genomic position.ref: The reference base/amino acid of the variant.alt: The alternative base/amino acid of the variant.coding: A flag indicating if the element is coding variant or not.
Per-variant summary of variant-producing guides (
tilingmode)guide_target_group: Aggregatedtarget_groupcolumn in the input sgRNA_info.csv file. All unique values of the guides that produced (filtered) edited alleles that includes this variant is listed.effective_edit_rate: The effective editing rate of the element. Calculated assum_over_guides(sum_over_alleles(per_guide_allele_editing_rate / # variants in the allele)).editing_guides: List of guides that edited the variant.per_guide_editing_rates: The per-guide editing rates of the variant.n_guides: The number of guides that edited the variant.n_coocc: The number of unique co-occurring variants that appeared together in any alleles that contains the variant.
Variant effect size: Use
mu_z_adjwhenever available, otherwisemu_z_scaled, otherwisemu_z.mu: The mean value of the variant effect size.mu_sd: The standard deviation of the mean value of the variant effect size.mu_z: The z-score of the mean value of the variant effect size.sd: The standard deviation of the phenotype induced by the variant.CI[0.025,0.975]: The 95% credible interval of the mean value of the variant effect size. Corresponds tomu_z_adjwhen available, otherwisemu_z_scaled, otherwisemu_z.[]_scaled: Above values scaled by negative control variants.[]_adj: Above values scaled by synonymous variants.
bean_sgRNA_result.[model_type].csv¶
name: sgRNA ID provided in thenamecolumn of the input.edit_rate: Effective editing ratesaccessibility: (Only if you have used--scale-by-acc) Accessibility signal that is used for scaling of the editing rate.scaled_edit_rate: (Only if you have used--scale-by-acc) Endogenous editing rate used for modeling, estimated by scaling reporter editing rate by accessibility signal[cond1]_[cond2].median_lfc: Raw LFC with pseudocount fed in with--guide-lfc-pseudocountargument (default 5).For
tilingmodevariants: Variants generated by this gRNAvariant_edit_rates: Editing rate of this gRNA for each variant it creates.
See the full output file description and example output here.
Full parameters¶
Run model on data.
usage: bean run [-h] [--uniform-edit] [--scale-by-acc]
[--acc-bw-path ACC_BW_PATH] [--acc-col ACC_COL]
[--outdir OUTDIR] [--result-suffix RESULT_SUFFIX] [--cuda]
[--fit-negctrl]
[--guide-lfc-pseudocount GUIDE_LFC_PSEUDOCOUNT]
[--dont-fit-noise]
[--dont-adjust-confidence-by-negative-control]
[--load-existing] [--save-raw] [--device DEVICE]
[--condition-col CONDITION_COL] [--time-col TIME_COL]
[--control-condition CONTROL_CONDITION]
[--plasmid-condition PLASMID_CONDITION]
[--replicate-col REPLICATE_COL] [--target-col TARGET_COL]
[--guide-activity-col GUIDE_ACTIVITY_COL]
[--sorting-bin-upper-quantile-col SORTING_BIN_UPPER_QUANTILE_COL]
[--sorting-bin-lower-quantile-col SORTING_BIN_LOWER_QUANTILE_COL]
[--sample-mask-col SAMPLE_MASK_COL]
[--negctrl-col NEGCTRL_COL]
[--negctrl-col-value NEGCTRL_COL_VALUE]
[--repguide-mask REPGUIDE_MASK]
[--allele-df-key ALLELE_DF_KEY]
[--splice-site-path SPLICE_SITE_PATH]
[--control-guide-tag CONTROL_GUIDE_TAG] [--n-iter N_ITER]
[--ignore-bcmatch] [--prior-params PRIOR_PARAMS] [--rep-pi]
[--const-pi] [--shrink-alpha]
[--exclude-control-condition-for-inference]
[--alpha-if-overdispersion-fitting-fails ALPHA_IF_OVERDISPERSION_FITTING_FAILS]
{sorting,survival} {variant,tiling} bdata_path
Positional Arguments¶
- selection
Possible choices: sorting, survival
Screen selection type whether cells are sorted based on continuous phenotype (‘sorting’) or proliferated based on their viability (‘survival’).
- library_design
Possible choices: variant, tiling
Library design type whether to run variant or tiling screen model. Variant library design assumes gRNA has specific target variant and bystander edits are ignored. Tiling library design considers all alleles generated by gRNA in reporter.
- bdata_path
Path of an ReporterScreen object
General run options¶
- --uniform-edit, -p
Assume uniform editing rate for all guides.
Default:
False- --scale-by-acc
Scale guide editing efficiency by the target loci accessibility
Default:
False- --acc-bw-path
Accessibility .bigWig file to be used to assign accessibility of guides.
- --acc-col
Column name in bdata.guides that specify raw ATAC-seq signal.
- --outdir, -o
Directory to save the run result.
Default:
'.'- --result-suffix
Suffix of the output files
Default:
''- --cuda
run on GPU
Default:
False- --fit-negctrl
Fit the shared negative control distribution to normalize the fitted parameters
Default:
False- --guide-lfc-pseudocount
LFC pseudocount that will be output as the bean_sgRNA_result.csv
Default:
5- --dont-fit-noise
Default:
False- --dont-adjust-confidence-by-negative-control
Do not adjust confidence by negative controls. Without this flag, variant mode will use negative control variants, and tiling mode will use the synonymous variants to adjust confidence of the result.
Default:
False- --load-existing
Load existing .pkl file if present.
Default:
False- --save-raw
Write .pkl file with raw input/output.
Default:
False- --device
Optionally use GPU if provided valid GPU device name (ex. cuda:0)
Input .h5ad formatting¶
- --condition-col
Column key in bdata.samples that describes experimental condition.
Default:
'condition'- --time-col
Column key in bdata.samples that describes time elapsed.
Default:
'time'- --control-condition
Comma-separated list of condition values in bdata.samples[condition_col] that indicates control experimental condition whose editing patterns will be used.
Default:
'bulk'- --plasmid-condition
For survival screens, condition label of the plasmid library, if included in the ReporterScreen.
Default:
'bulk'- --replicate-col
Column key in bdata.samples that describes experimental replicates.
Default:
'replicate'- --target-col
Column key in bdata.guides that describes the target element of each guide.
Default:
'target'- --guide-activity-col, -a
Column in ReporterScreen.guides DataFrame showing the editing rate estimated via external tools
- --sorting-bin-upper-quantile-col, -uq
Column name with upper quantile values of each sorting bin in [Reporter]Screen.samples (or AnnData.var)
Default:
'upper_quantile'- --sorting-bin-lower-quantile-col, -lq
Column name with lower quantile values of each sorting bin in [Reporter]Screen.samples (or AnnData var)
Default:
'lower_quantile'- --sample-mask-col
Name of the column indicating the sample mask in [Reporter]Screen.samples (or AnnData.var). Sample is ignored if the value in this column is 0. This can be used to mask out low-quality samples. If you don’t want to mask samples out, provide –sample-mask-col=’’.
Default:
'mask'- --negctrl-col
Column in bdata.obs specifying if a guide is negative control. If the bdata.guides[negctrl_col].lower() == negctrl_col_value, it is treated as negative control guide.
Default:
'target_group'- --negctrl-col-value
Column value in bdata.guides specifying if a guide is negative control. If the bdata.guides[negctrl_col].lower() == negctrl_col_value, it is treated as negative control guide.
Default:
'negctrl'- --repguide-mask
n_replicate x n_guide mask to mask the outlier guides. screen.uns[repguide_mask] will be used.
Default:
repguide_mask- --allele-df-key
screen.uns[allele_df_key] will be used as the allele count.
- --splice-site-path
Path to splicing site
- --control-guide-tag
If this string is in guide name, treat each guide separately not to mix the position. Used for negative controls.
Advanced arguments for model fitting¶
- --n-iter
# of SVI steps taken for inference.
Default:
2000- --ignore-bcmatch
If provided, even if the screen object has .X_bcmatch, ignore the count when fitting.
Default:
False- --prior-params
Path to the .pkl file with the dictionary containing prior parameters. Useful if your screens are batched into disjoint pool of gRNA libraries. Currently supports mu_loc, mu_scale, sd_loc, sd_scale for sorting screens and mu_loc, mu_scale, initial_abundance for survival screens.
- --rep-pi, -r
Fit replicate specific scaling factor. Recommended to set as True if you expect variable editing activity across biological replicates.
Default:
False- --const-pi
Use constant pi provided in –guide-activity-col (instead of fitting from reporter data)
Default:
False- --shrink-alpha
Instead of using the trend-fitted alpha values, use estimated alpha values for each gRNA that are shrunk towards the fitted trend.
Default:
False- --exclude-control-condition-for-inference, -ec
Exclude control conditions for inference. Currently only supported for survival screens.
Default:
False- --alpha-if-overdispersion-fitting-fails, -af
Comma-separated regression coefficient (b0, b1) of log(a0) ~ log(q) that will be used if fitting dispersion on the data fails.
