bean filter¶
filter: Filtering (and optionally translating) alleles¶
As tiling mode of bean run accounts for any robustly observed alleles, bean filter filters for such alleles.
bean filter my_sorting_screen_masked.h5ad \
-o my_sorting_screen_filtered.h5ad `# Output file path` \
Output¶
Above command produces
my_sorting_screen_filtered.h5adwith filtered alleles stored in.uns,my_sorting_screen_filtered.filtered_allele_stats.pdf, andmy_sorting_screen_filtered.filter_log.txtthat report allele count stats in each filtering step.
You may want to adjust the flitering parameters to obtain optimal balance between # guides per variant & # variants that are scored. See example outputs of filtering step here.
Translating alleles¶
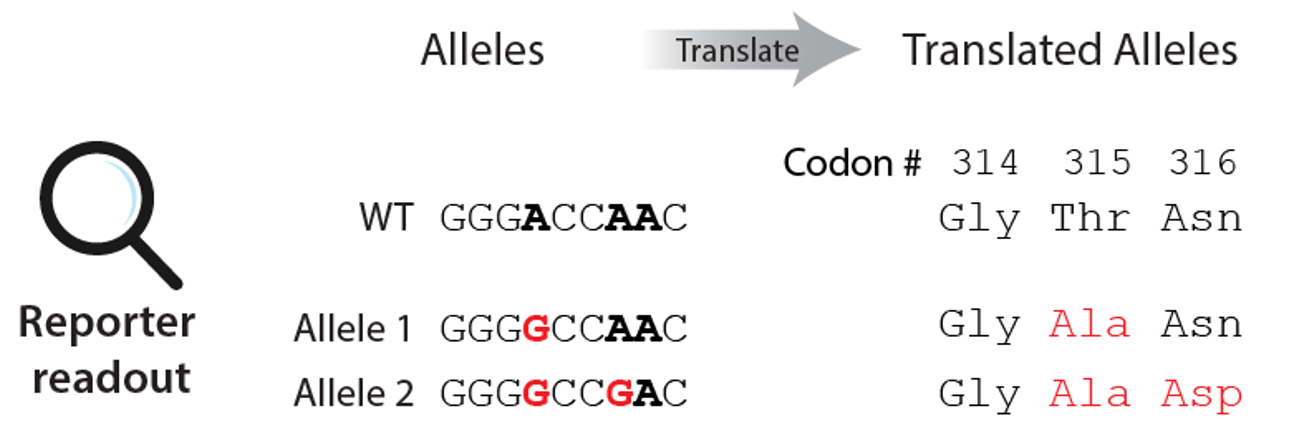
If you want to obtain amino acid level variant for coding sequence tiling screens, provide coding sequence positions which variants occuring within the coding sequence will be translated. This is optional, but **highly recommended* to increase per-(coding)variant support.*
bean filter my_sorting_screen.h5ad \
-o my_sorting_screen_masked.h5ad \
--translate `# Translate coding variants` \
[ --translate-gene-name GENE_SYMBOL OR
--translate-genes-list path_to_gene_names_file.txt OR
--translate-fasta gene_exon.fa, OR
--translate-fastas-csv gene_exon_fas.csv]
When library covers a single gene, do either of the following:
Feed
--translate-gene-name GENE_SYMBOLif yourgenomic_poscolumn ofsgRNA_info_tblis compatible with MANE transcript‘s reference genome. (Per 10/23/2023, GRCh38). This will automatically load the exon positions based on MANE transcript annotation.To use your custom coding sequence and exon positions, feed
--translate-fasta gene_exon.faargument wheregene_exon.fais the FASTA file with entries of exons. See full details here.
When library covers multiple genes, do either of the following:
Feed
--translate-genes-list path_to_gene_names_file.txtwherepath_to_gene_names_file.txtis file with one gene symbol per line.Feed
--translate-fastas-csv gene_exon_fas.csvwheregene_exon_fas.csvis the csv file with linesgene_id,gene_exon_fasta_pathwithout header. Each FASTA file ingene_exon_fasta_pathis formatted as the single-gene FASTA file.
Translation will keep the variants outside the coding sequence as nucleotide-level variants, while aggregating variants leading to the same coding sequence variants.
Full parameters¶
Filter alleles based on edit position in spacer and frequency across samples.
usage: bean filter [-h] [--output-prefix OUTPUT_PREFIX]
[--plasmid-path PLASMID_PATH]
[--reporter-length REPORTER_LENGTH]
[--reporter-right-flank-length REPORTER_RIGHT_FLANK_LENGTH]
[--edit-start-pos EDIT_START_POS]
[--edit-end-pos EDIT_END_POS]
[--jaccard-threshold JACCARD_THRESHOLD] [--filter-spacer]
[--filter-window] [--keep-indels]
[--filter-target-basechange] [--translate]
[--translate-fasta TRANSLATE_FASTA]
[--translate-fastas-csv TRANSLATE_FASTAS_CSV]
[--translate-gene TRANSLATE_GENE]
[--translate-genes-list TRANSLATE_GENES_LIST]
[--filter-allele-proportion FILTER_ALLELE_PROPORTION]
[--filter-allele-count FILTER_ALLELE_COUNT]
[--filter-sample-proportion FILTER_SAMPLE_PROPORTION]
[--load-tmp]
bdata_path
Positional Arguments¶
- bdata_path
Input ReporterScreen file of which allele will be filtered out.
Named Arguments¶
- --output-prefix, -o
Output prefix for log and ReporterScreen file with allele assignment
- --plasmid-path, -p
Plasmid ReporterScreen object path. If provided, alleles are filtered based on if a nucleotide edit is more significantly enriched in sample compared to the plasmid data. Negative control data where no edit is expected can be fed in instead of plasmid library.
- --reporter-length
Length of reporter sequence in the construct.
- --reporter-right-flank-length
Length of the right-flanking nucleotides of protospacer in the reporter.
- --edit-start-pos, -s
0-based start posiiton (inclusive) of edit relative to the start of guide spacer.
Default:
2- --edit-end-pos, -e
0-based end position (exclusive) of edit relative to the start of guide spacer.
Default:
7- --jaccard-threshold, -j
Jaccard Index threshold when the alleles are mapped to the most similar alleles. In each filtering step, allele counts of filtered out alleles will be mapped to the most similar allele only if they have Jaccard Index of shared edit higher than this threshold.
Default:
0.3- --filter-spacer
Only consider edit within protospacer positions of reporter.
Default:
False- --filter-window, -w
Only consider edit within window provided by (edit-start-pos, edit-end-pos). If this flag is not provided, –edit-start-pos and –edit-end-pos flags are ignored.
Default:
False- --keep-indels, -i
Include indels.
Default:
False- --filter-target-basechange, -b
Only consider target edit (stored in bdata.uns[‘target_base_changes’])
Default:
False- --translate, -t
Translate alleles
Default:
False- --translate-fasta, -f
fasta file path with exon positions. If not provided, LDLR hg19 coordinates will be used.
- --translate-fastas-csv, -fs
.csv with two columns with gene IDs and FASTA file path corresponding to each gene.
- --translate-gene, -g
Gene symbol if a gene is tiled. If not provided, LDLR hg19 coordinates will be used.
- --translate-genes-list, -gs
File with gene symbols, one per line, if multiple genes are tiled.
- --filter-allele-proportion, -ap
If provided, alleles that exceed filter_allele_proportion in filter-sample-proportion will be retained.
Default:
0.05- --filter-allele-count, -ac
If provided, alleles that exceed filter_allele_proportion AND filter_allele_count in filter-sample-proportion will be retained.
Default:
5- --filter-sample-proportion, -sp
If filter_allele_proportion is provided, alleles that exceed filter_allele_proportion in filter-sample-proportion will be retained.
Default:
0.2- --load-tmp
Load temporary file and work from there.
Default:
False
bean get-splice-sites¶
Getting splice sites¶
We provide the utility script to obtain the splice sites if you use the MANE transcript with gene symbol (--translate-gene-name GENE_SYMBOL or --translate-genes-list path_to_gene_names_file.txt).
bean get-splice-sites LDLR A LDLR_splice_sites.csv --gene-name
Parameters¶
usage: Get splice site position from exon fasta and target editing base.
Positional Arguments¶
- exon_fa_path
File path to fasta file with exon position information.
- edited_base
Edited base, either A or C.
- output_path
output path of the splice site csv file.
Named Arguments¶
- --gene-name
File path to fasta file with exon position information.
Default:
False
