Evaluating Peak-to-Gene Predictions with Experimental Validation#
This tutorial demonstrates how to evaluate and compare peak-to-gene predictions from different computational methods using experimental validation data. We’ll use Hi-C and ChIA-PET datasets as ground truth to assess the quality of predicted regulatory relationships.
Overview#
Computational prediction of peak-to-gene links is a fundamental challenge in single-cell genomics. To objectively compare different methods, we need experimental validation datasets that capture true regulatory relationships. This tutorial shows how to:
Load experimental validation data (Hi-C and ChIA-PET)
Compare computational predictions against experimental evidence
Calculate enrichment metrics to quantify method performance
Visualize comparative results across multiple methods
Experimental Validation Datasets#
Hi-C Data:#
Technology: Chromosome conformation capture (3C-based)
Information: Genome-wide chromatin interactions
Resolution: Identifies both promoter-enhancer and structural interactions
Strength: Comprehensive, unbiased interaction mapping
ChIA-PET Data:#
Technology: Chromatin interaction analysis by paired-end tag sequencing
Information: Protein-mediated chromatin interactions
Resolution: Higher specificity for transcription factor-mediated loops
Strength: Identifies functional regulatory interactions
Evaluation Strategy:#
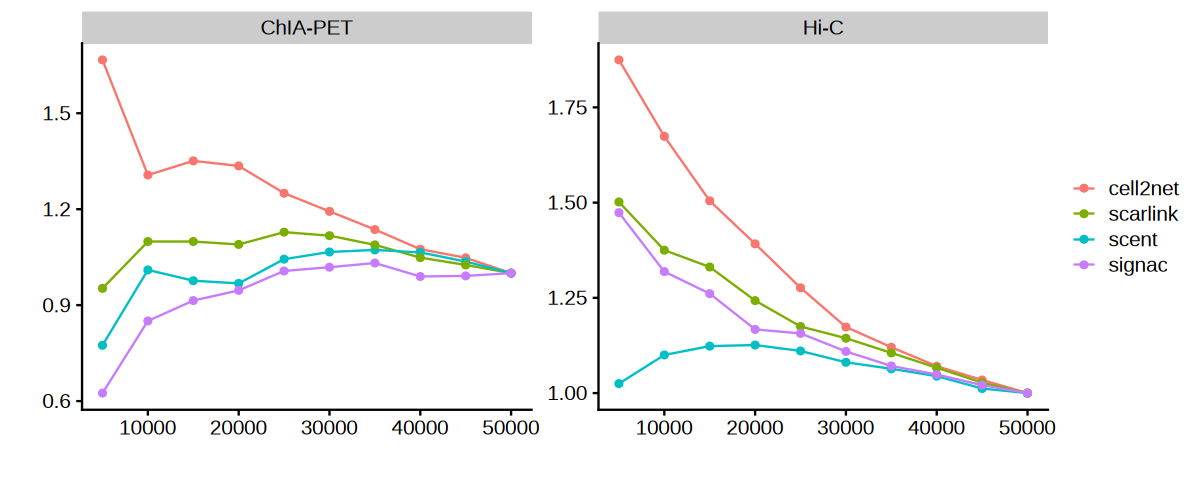
We use odds ratio analysis to measure how well each method’s top predictions are enriched for experimentally validated interactions compared to random expectations. Higher odds ratios indicate better performance in identifying true regulatory relationships.
suppressMessages(library(cicero))
suppressMessages(library(ggplot2))
suppressMessages(library(cowplot))
suppressMessages(library(gridExtra))
suppressMessages(library(dplyr))
Setup Input Directory#
Define the path to processed peak-to-gene predictions from the previous tutorial (05_process_p2g).
in_dir <- "./05_process_p2g"
Load Experimental Validation Data#
Import Hi-C and ChIA-PET datasets that serve as ground truth for evaluating computational peak-to-gene predictions.
Hi-C Data (ENCFF256ZMD):#
Source: ENCODE K562 Hi-C experiment
Technology: Genome-wide chromatin interaction mapping
Format: BEDPE (paired genomic intervals)
Coverage: Comprehensive interaction landscape
ChIA-PET Data (ENCFF607PZX):#
Source: ENCODE K562 ChIA-PET experiment
Technology: Protein-mediated chromatin interactions
Format: BEDPE (paired genomic intervals)
Specificity: Transcription factor-mediated loops
Both datasets are processed to create standardized coordinate formats matching our computational predictions.
# read Hi-C and ChIA-PET data
df_hic <- read.table("../../../../data/K562_HiC/ENCFF256ZMD.bedpe",
sep = "\t",
header = FALSE,
stringsAsFactors = FALSE)
colnames(df_hic)[1:6] <- c("chr1", "start1", "end1",
"chr2", "start2", "end2")
df_hic$Peak1 <- paste(df_hic$chr1, df_hic$start1, df_hic$end1, sep = "_")
df_hic$Peak2 <- paste(df_hic$chr2, df_hic$start2, df_hic$end2, sep = "_")
df_hic <- df_hic[c("Peak1", "Peak2")]
df_chiapet <- read.table("../../../../data/K562_ChIAPET/ENCFF607PZX.bedpe",
sep = "\t",
header = FALSE,
stringsAsFactors = FALSE)
colnames(df_chiapet)[1:6] <- c("chr1", "start1", "end1",
"chr2", "start2", "end2")
df_chiapet$Peak1 <- paste(df_chiapet$chr1, df_chiapet$start1, df_chiapet$end1, sep = "_")
df_chiapet$Peak2 <- paste(df_chiapet$chr2, df_chiapet$start2, df_chiapet$end2, sep = "_")
df_chiapet <- df_chiapet[c("Peak1", "Peak2")]
Define Evaluation Function#
Create a function to compute odds ratios that measure how well computational predictions are enriched for experimentally validated interactions.
Methodology:#
Compare predictions with validation data using genomic overlap (±100bp tolerance)
Calculate baseline proportion from top 50,000 predictions
Compute odds ratios for different prediction set sizes (5K to 50K)
Return enrichment metrics for performance assessment
Key Metrics:#
Proportion linked: Fraction of predictions supported by experimental data
Odds ratio: Enrichment relative to baseline expectation
Progressive evaluation: Performance across different stringency levels
Statistical Rationale:#
Higher odds ratios indicate that a method’s top predictions are more enriched for true regulatory relationships compared to random expectations. This provides an objective measure of method performance.
compute_ratio <- function(df_pred, df_true){
prop_list <- list()
ratio_list <- list()
n_links_list <- list()
df_pred$isLinked <- compare_connections(df_pred, df_true, maxgap=100)
df_top_50k <- df_pred %>%
arrange(desc(score)) %>%
slice_head(n = 50000)
odds_b = sum(df_top_50k$isLinked) / nrow(df_top_50k)
for(top_n in c(5000, 10000, 15000, 20000, 25000, 30000, 35000, 40000, 45000, 50000)){
df_top <- df_pred %>%
arrange(desc(score)) %>%
slice_head(n = top_n)
odds_a = sum(df_top$isLinked) / nrow(df_top)
odds_ratio = odds_a / odds_b
# Store results
n_links_list <- append(n_links_list, top_n)
prop_list <- append(prop_list, odds_a)
ratio_list <- append(ratio_list, odds_ratio)
}
df_res <- data.frame(
n_links = unlist(n_links_list),
prop_linked = unlist(prop_list),
odds_ratio = unlist(ratio_list)
)
return(df_res)
}
Evaluate All Methods Against Validation Data#
Systematically evaluate each computational method (Cell2Net, SCENT, SCARlink, Signac) against both experimental validation datasets (Hi-C and ChIA-PET).
Process:#
Load predictions from each method (processed in tutorial 05)
Apply evaluation function to compute odds ratios
Compare against both datasets for comprehensive assessment
Combine results into unified dataframe for visualization
Track method performance across different prediction set sizes
Output Structure:#
n_links: Number of top predictions evaluated
prop_linked: Proportion supported by experimental data
odds_ratio: Enrichment relative to baseline
method: Computational approach name
data: Validation dataset type (Hi-C or ChIA-PET)
df_final <- lapply(c("cell2net", "scent", "scarlink", "signac"), function(method) {
df_pred <- read.csv(glue::glue("{in_dir}/{method}.csv"), row.names = 1)
df_hic_res <- compute_ratio(df_pred, df_hic)
df_chiapet_res <- compute_ratio(df_pred, df_chiapet)
df_hic_res$method <- method
df_chiapet_res$method <- method
df_hic_res$data <- "Hi-C"
df_chiapet_res$data <- "ChIA-PET"
df <- rbind(df_hic_res, df_chiapet_res)
return(df)
} ) %>%
bind_rows()
Let’s examine the structure of our evaluation results:
head(df_final)
| n_links | prop_linked | odds_ratio | method | data | |
|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <chr> | <chr> | |
| 1 | 5000 | 0.05980000 | 1.874608 | cell2net | Hi-C |
| 2 | 10000 | 0.05340000 | 1.673981 | cell2net | Hi-C |
| 3 | 15000 | 0.04800000 | 1.504702 | cell2net | Hi-C |
| 4 | 20000 | 0.04440000 | 1.391850 | cell2net | Hi-C |
| 5 | 25000 | 0.04072000 | 1.276489 | cell2net | Hi-C |
| 6 | 30000 | 0.03743333 | 1.173459 | cell2net | Hi-C |
Visualize Method Performance Comparison#
Create comprehensive plots showing how each method performs against experimental validation data.
options(repr.plot.height = 4, repr.plot.width = 10)
p <- ggplot(data = df_final, aes(x = n_links, y = odds_ratio)) +
geom_line(aes(color = method)) +
geom_point(aes(color = method)) +
facet_wrap(~data, scales = "free") +
xlab("") + ylab("") +
theme_cowplot() +
theme(legend.title = element_blank())
print(p)